Tutorial 7: Morphology-Aware Analysis of Subcellular Protein Localization in Large Datasets (dCellAligner-OT)
The Fused Gromov-Wasserstein mapping between two cells with 1,000 points takes around 3 s to compute on a standard desktop computer, whereas the optimal transport (OT) distance between the two mapped distributions takes around 18 ms. Although the number of Fused Gromov-Wasserstein mapping computations scales linearly with the number of cells and the number of OT distance computations between mapped distributions scales quadratically, this can result in long runtimes for datasets with hundreds of thousands of cells.
For these large datasets, CAJAL provides a deep learning framework, dCellAligner-OT, to reduce the required computation. This approach allows users to compute CellAligner mappings and OT distances for only a subset of cells, then train a deep learning model to predict the mappings and distances for the remaining cells.
We will demonstrate this approach on a dataset of 16,787 neurons with simulated subcellular protein distributions. For this analysis, we assume that the image data has already been processed into CellAligner_Cell objects (as described in Tutorial 6), which can be downloaded from this link.
[ ]:
import os
from cajal.subcellular import *
from cajal.subcellular_dl import *
data_path = '/workspaces/CellAligner/sim_neuron_cell_objects/cell_objects/' # Path to directory containing cell object files
cell_object_paths = [os.path.join(data_path, fname) for fname in os.listdir(data_path)]
anchor_ind = 658 # index of anchor cell (which other cells are mapped to)
anchor_cell_obj_path = cell_object_paths[anchor_ind]
with open('/workspaces/CellAligner/sim_neuron_cell_objects/anchor_658_mapped_distbs.pickle', 'rb') as file:
mapped_distbs = pickle.load(file)
First, we convert the CellAligne_Cell objects and their mapped subcellular protein distributions into cell-specific images that can be used to the dCellAligner-OT model. The make_NN_training_data function creates two directories, cell_images and mapped_cell_images, which store the original and mapped cell images, respectively. It also generates a configuration file, cell_image_processing.json, containing the image-processing parameters used for alignment, centering, resizing,
and related steps. This file must be referenced when applying dCellAligner-OT to new CellAligner_Cell objects so that image processing remains consistent.
[ ]:
cell_image_path = '/workspaces/CellAligner/sim_neuron_cell_objects/' # Path to directory where generated cell images will be saved
make_NN_training_data(save_path=cell_image_path,
cell_objects=cell_object_paths,
reference_cell_object=anchor_cell_obj_path,
mapped_channel_distributions=mapped_distbs[0], # using the mapped protein distribution
channel='protein', # this should match the mapped distributions used
center='nucleus', # center='cell' when using Fused GW mappings, center='nucleus' when using Unbalanced Fused GW mappings
shape=(256,256), # shape of the output cell images
rescale=False) # rescale=True when using Fused GW mappings, rescale=False when using Unbalanced Fused GW mappings
To reduce overfitting, we split the data into training, validation, and test sets. The dCellAligner model does not need to be trained on all possible pairs of training cells, so we compute OT distances between mapped protein distributions for only a subset of cell pairs. In practice, we have observed good performance when training on approximately 10,000 cells and 30,000 cell pairs.
[17]:
# Generate train/val/test dataset cell pairs
train_pairs, val_pairs, test_pairs = generate_dataset_split_pairs(indices=list(range(len(cell_object_paths))),
n_pairs=[35000, 10000, 5000], # number of cell pairs in train/val/test sets
proportions=[0.7, 0.2, 0.1]) # proportion of cells in train/val/test sets
# Store unique indices in each set
train_inds = np.unique(train_pairs)
val_inds = np.unique(val_pairs)
test_inds = np.unique(test_pairs)
# Compute GW-mapped OT distances for all pairs in train/val/test sets
train_ot_dists = gw_mapped_ot_pairwise_parallel(cell_object_paths[anchor_ind], mapped_distbs,
num_processes=12, chunksize=20, index_pairs=train_pairs)[0]
val_ot_dists = gw_mapped_ot_pairwise_parallel(cell_object_paths[anchor_ind], mapped_distbs,
num_processes=12, chunksize=20, index_pairs=val_pairs)[0]
test_ot_dists = gw_mapped_ot_pairwise_parallel(cell_object_paths[anchor_ind], mapped_distbs,
num_processes=12, chunksize=20, index_pairs=test_pairs)[0]
# Create PairedDataset objects for train/val/test sets for training dCellAligner-OT
train_data = PairedDataset(
image_dir = cell_image_path,
mapped_image_dir = cell_image_path,
distances = train_ot_dists.astype('float32'),
image_pairs = train_pairs,
transform = transforms.Compose([transforms.ToImage(),
transforms.ToDtype(torch.float32)]),
)
val_data = PairedDataset(
image_dir = cell_image_path,
mapped_image_dir = cell_image_path,
distances = val_ot_dists.astype('float32'),
image_pairs = val_pairs,
transform = transforms.Compose([transforms.ToImage(),
transforms.ToDtype(torch.float32)]),
)
test_data = PairedDataset(
image_dir = cell_image_path,
mapped_image_dir = cell_image_path,
distances = test_ot_dists.astype('float32'),
image_pairs = test_pairs,
transform = transforms.Compose([transforms.ToImage(),
transforms.ToDtype(torch.float32)]),
)
Computing pairwise OT distances:
100%|██████████| 35000/35000 [07:26<00:00, 78.44it/s]
Computing pairwise OT distances:
100%|██████████| 10000/10000 [03:16<00:00, 50.90it/s]
Computing pairwise OT distances:
100%|██████████| 5000/5000 [01:43<00:00, 48.46it/s]
We initialize the dCellAligner-OT model and begin the two-stage training process. During the first stage, or pretraining, the model learns to approximate the mapping operation. More specifically, for each cell, the model learns to predict the subcellular protein distribution after mapping to the anchor cell.
In our benchmark, dCellAligner-OT pretraining took approximately 24 hours on an NVIDIA RTX 4500 Ada GPU.
[ ]:
model = dCellAlignerNetwork(embedding_size=50, image_size=image_shape[0])
model = pretrain_model(train_data, model, dataset_name='sim_neuron', batch_size=8, epochs=50, lr=1e-3,
device='cuda', save_path='/workspaces/CellAligner/sim_neuron_cell_objects/pretrained_model/', return_model=True) # Replace with path to save the pretrained model
Next, during training, the model learns to extract features that preserve CellAligner-OT distances between cells in the latent feature space, while continuing to predict the mapped protein distributions.
The model is optimized with respect to two main loss components. The distance loss measures how well CellAligner-OT distances are preserved in the model’s latent feature space, whereas the reconstruction loss measures how accurately the model predicts the mapped protein distributions. The dist_weight parameter controls the relative weighting of these loss components during training. Ideally, the contributions of the two losses, which can be inspected by setting
show_loss_components = True, should be of similar order of magnitude.
To reduce overfitting, we apply L1 regularization, controlled by weight_decay, and L2 regularization, controlled by sparsity_weight and sparsity_target. If the dCellAligner-OT model is overfitting, users can try increasing weight_decay or sparsity_weight, or decreasing sparsity_target, to regularize the model more strongly.
In our benchmark, dCellAligner-OT training took approximately 48 hours on an NVIDIA RTX 4500 Ada GPU.
[ ]:
# Train the dCellAligner-OT with the prepared datasets
models, train_losses, val_losses = train_dCellAligner(
train_data, val_data, test_data,
save_path='/workspaces/CellAligner/sim_neuron_cell_objects/fully_trained_model/', # Replace with path to save the fully trained model
dataset_name='sim_neuron',
embedding_size=50, # 50-dimensional embeddings
image_shape=(256, 256), # Input image shape
device='cuda', # Use GPU if available
batch_size=8, # Batch size for training
epochs=25, # Number of epochs
learning_rate=0.001, # Adam learning rate
dist_weight=0.1, # Distance weight (vs reconstruction loss)
early_stopping=False, # Disable early stopping
weight_decay=1e-4, # L2 regularization weight
lr_gamma=0.95, # Learning rate decay factor
sparsity_weight=1e-3, # Sparsity weight for the embedding loss
sparsity_target=0.1, # Target sparsity for the embedding loss
pretrained_path="/workspaces/CellAligner/sim_neuron_cell_objects/pretrained_model/sim_neuron_pretrained_best.pth"
)
The train_dCellAligner function saves two model checkpoints: <dataset_name>_best.pth, corresponding to the best validation-set performance, and <dataset_name>_final.pth, corresponding to the model after the final training epoch. Here, we load the best-performing model based on validation loss.
[18]:
# load best model
model = load_dCellAligner_model('/workspaces/CellAligner/sim_neuron_cell_objects/fully_trained_model/sim_neuron_best.pth')
/opt/conda/lib/python3.12/site-packages/cajal/subcellular_dl.py:1276: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
checkpoint = torch.load(checkpoint_path, map_location=device)
/opt/conda/lib/python3.12/site-packages/torchvision/models/_utils.py:208: UserWarning: The parameter 'pretrained' is deprecated since 0.13 and may be removed in the future, please use 'weights' instead.
warnings.warn(
/opt/conda/lib/python3.12/site-packages/torchvision/models/_utils.py:223: UserWarning: Arguments other than a weight enum or `None` for 'weights' are deprecated since 0.13 and may be removed in the future. The current behavior is equivalent to passing `weights=EfficientNet_B4_Weights.IMAGENET1K_V1`. You can also use `weights=EfficientNet_B4_Weights.DEFAULT` to get the most up-to-date weights.
warnings.warn(msg)
Downloading: "https://download.pytorch.org/models/efficientnet_b4_rwightman-23ab8bcd.pth" to /home/jovyan/.cache/torch/hub/checkpoints/efficientnet_b4_rwightman-23ab8bcd.pth
100%|██████████| 74.5M/74.5M [00:09<00:00, 8.55MB/s]
Loaded Deep CellAligner model from /workspaces/CellAligner/sim_neuron_cell_objects/fully_trained_model/sim_neuron_best.pth
Config: {'input_channels': 3, 'embedding_size': 50, 'image_size': 256}
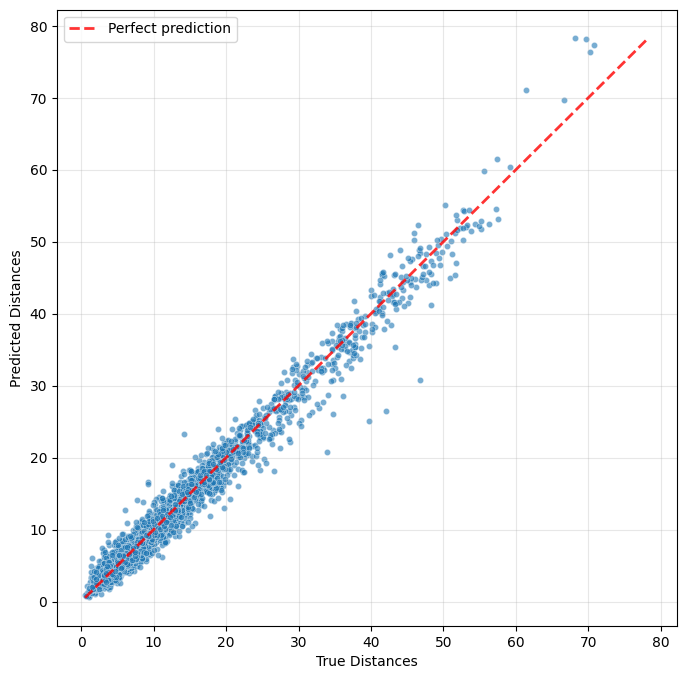
To evaluate model performance, we consider two criteria: how well the dCellAligner-OT feature space preserves the true CellAligner-OT distances, and how accurately the model predicts the mapped protein distributions.
[20]:
plot_distance_predictions(model, test_data)
Extracting embeddings for 1676 unique images...
Extracting embeddings: 100%|██████████| 27/27 [15:59<00:00, 35.52s/it]
Computing distances for 5000 pairs...
Computing pairwise distances: 100%|██████████| 5000/5000 [00:00<00:00, 101063.18it/s]
[23]:
plot_reconstruction_comparison(model, test_data)

Finally, the trained model can be used to extract features that capture variation in subcellular protein localization in datasets of arbitrary size, without requiring additional training or OT distance computations. This feature space can then be used for the same downstream analyses as the CellAligner-OT localization space, including clustering and visualization.
[29]:
import umap as umap
import plotly.express
# load cell metadata
cell_metadata = pd.read_csv('/workspaces/CellAligner/sim_neuron_cell_objects/cell_metadata.csv', index_col=0)
embeddings = extract_embeddings(model, test_data)
# Compute UMAP representation
reducer = umap.UMAP(random_state=1)
umap_coords = reducer.fit_transform(embeddings)
plotly.express.scatter(x=umap_coords[:,0],
y=umap_coords[:,1],
template="simple_white",
hover_name=cell_metadata.iloc[np.unique(test_pairs)]['hpa_crop_id'],
color=cell_metadata.iloc[np.unique(test_pairs)]['hpa_locations'])
Extracting embeddings: 100%|██████████| 27/27 [13:04<00:00, 29.04s/it]
/opt/conda/lib/python3.12/site-packages/umap/umap_.py:1952: UserWarning: n_jobs value 1 overridden to 1 by setting random_state. Use no seed for parallelism.
warn(
The model evaluations above used cell images for which anchor-cell mappings and OT distances had already been computed. After training, however, dCellAligner-OT can be applied directly to new CellAligner_Cell objects to infer anchor-cell mappings and estimate CellAligner-OT distances much more efficiently.
[26]:
# Select random cells to visualize
np.random.seed(0)
cells_to_plot = np.random.choice(len(cell_object_paths), size=3, replace=False)
new_cell_object_paths = [cell_object_paths[i] for i in cells_to_plot]
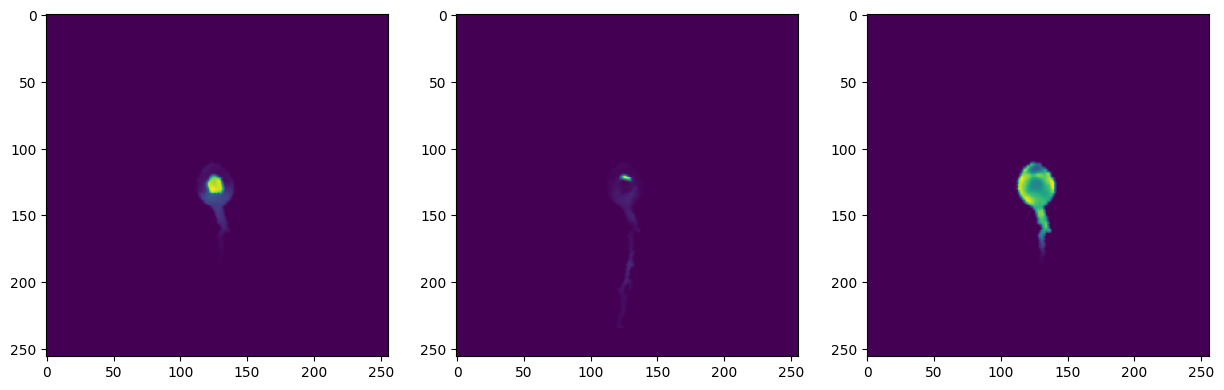
# Use dCellAligner model to map cells to anchor cell
mapped_images = deep_map_to_anchor_cell(model, new_cell_object_paths, process_info_path=os.path.join(cell_image_path, 'cell_image_processing.json'), channel='protein')
fig, axes = plt.subplots(1, len(new_cell_object_paths), figsize=(15, 5))
for ax, i in zip(axes, range(len(new_cell_object_paths))):
ax.imshow(mapped_images[i])
plt.show()
Mapping cells: 100%|██████████| 1/1 [00:04<00:00, 4.09s/it]
[28]:
# Compute pairwise distances for CellAligner_Cell objects
distances = predict_distances(model, new_cell_object_paths, channel='protein', process_info_path=os.path.join(cell_image_path, 'cell_image_processing.json'))
print(distances)
Extracting embeddings for 3 images...
Extracting embeddings: 100%|██████████| 1/1 [00:03<00:00, 3.41s/it]
Computing distance matrix for 3 cells:
[[ 0. 14.308745 4.5295916]
[14.308745 0. 13.887374 ]
[ 4.5295916 13.887374 0. ]]